ヘマンジオルの第Ⅱ/Ⅲ相臨床試験(海外データ)
試験概要
目的
全身治療が必要な増殖期の乳児血管腫患者を対象として、プロプラノロール経口液剤の4つの投与群から最適な用量及び投与期間を確認し、24週後における有効性と安全性をプラセボを対照に評価する。
試験デザイン
ランダム化、二重盲検、プラセボ対照、反復投与、多施設共同、アダプティブ試験※
アダプティブ試験:
試験実施中に、試験デザインを変更したり、試験の中止あるいは継続の判断をしたりするデザイン(アダプティブデザイン)に基づいて実施された試験
対象
乳児血管腫患者
【選択基準】
- 長径が1.5cm以上の増殖期の乳児血管腫を有し、全身治療が必要な患者
- 登録時に生後35~150日の患者
【主な除外基準】
- 先天性血管腫
- 以下に該当する乳児血管腫
- 生命を脅かすもの
- 機能を脅かす(視力障害、呼吸障害等)もの
- 痛みを伴い、通常の創傷治療で効果がない潰瘍を形成したもの
- 早産児で正期産に該当する日齢に達していない
- Kasabach-Merritt症候群
- 気管支喘息、気管支痙攣
- 低血糖(血糖値40mg/dL未満又はそのおそれのある患者)
- 低血圧(収縮期50mmHg未満、拡張期30mmHg未満)
- 徐脈(80回/分未満)
- コントロール不良の心不全
方法
本試験は以下のステージ1とステージ2から構成される。ステージ1は用量設定試験(第Ⅱ相臨床試験)、ステージ2までは有効性評価試験(第Ⅲ相臨床試験)に相当する。但し、各投与群の症例割付けはステージ1開始時に行った。最初の190例(ステージ1)が24週間の投与を完了するか、投与を中止した時点で、中間解析を実施した。
<ステージ1(190例)>
プロプラノロール経口液剤の4つの投与群をプラセボ群と比較した。
<ステージ2(270例)>
中間解析結果より適切な用法・用量と判断された3mg/kg/日 24週群とプラセボ群を有効性解析対象とした。なお、中間解析結果をもとにステージ2の解析方法を決定するまでは、4つの投与群及びプラセボ群への患者の割付けを継続し、適切な用法・用量と判断されなかった3つの投与群は有効性解析対象外とし、安全性解析対象とした。
【投与方法】
プロプラノロール経口液剤又はプラセボ(投与液量0.4mL/kgとなるように濃度を調整)を以下の①~⑤のいずれかの用量及び投与期間で、1日2回(朝・夜)、食事(授乳)の前後に経口投与した。
- ①プロプラノロール換算 1mg/kg/日の経口液剤を12週間投与(以下、1mg/kg/日・12週群)(13~24週はプラセボ投与)
- ②プロプラノロール換算 1mg/kg/日の経口液剤を24週間投与(以下、1mg/kg/日・24週群)
- ③プロプラノロール換算 3mg/kg/日の経口液剤を12週間投与(以下、3mg/kg/日・12週群)(13~24週はプラセボ投与)
- ④プロプラノロール換算 3mg/kg/日の経口液剤を24週間投与(以下、3mg/kg/日・24週群)
- ⑤プラセボを24週間投与(以下、プラセボ群)
3mg/kg/日投与群では漸増期間を設け、投与開始日~投与7日目に1mg/kg/日、投与8日目~投与14日目に2mg/kg/日、投与15日目以降に3mg/kg/日を投与した。残りの投与群でも擬似的な漸増期間を設けた。
初回の投与は医療機関で行った。また、投与開始日及び増量時(投与8日目及び投与15日目)には、投与前後の所定の時点にバイタルサイン(体温、心拍数、血圧、呼吸数)、肺音聴診、血糖値、心電図を測定・検査して、安全性を確認した。
評価項目
【主要評価項目】
24週後における乳児血管腫に対する有効率(血管腫が「治癒又はほぼ治癒」した割合)
【副次評価項目】
- 12週後における乳児血管腫に対する有効率
- 前回来院時からの乳児血管腫の改善率
- 乳児血管腫が改善するまでの期間
- 12週後、24週後における乳児血管腫の面積、長径、色の変化量
【安全性】
有害事象、臨床検査値、診察・理学的検査、バイタルサイン、血糖値(pin-prick)、心電図、神経発達の評価
解析方法
【主要評価項目】
Poschらが報告した中間解析時に1つの治療法を選択する検証的アダプティブデザインの手法を用いて、選択された治療法の優越性を検証する。有意水準は片側0.005とした。
主解析(ステージ1+ステージ2)
ステージ1の結果から、3mg/kg/日・24週群について閉検定手順(Simes法)を用いて調整P 値を算出した。3mg/kg/日・24週群のP 値は、閉検定手順による調整P 値とステージ2の片側Z検定のP 値を用いたPoschらの統合検定法により算出した。
判定基準
各来院ごとに標準的な手順に従って正面及び側面から撮影された各1枚のデジタル写真を用いて事前にトレーニングを受けた効果判定委員が、盲検下で患者内比較により下記の定性的な中央判定を行った。
- 乳児血管腫に対する有効判定:12週後、24週後及び中止(来院)日
投与開始日の写真と比較し、乳児血管腫の状態を「有効(治癒又はほぼ治癒)」「無効」の2段階で評価した。
「ほぼ治癒」の定義は「毛細血管拡張、紅斑、皮膚肥厚、軟組織の腫脹、解剖学的な境界の歪みのすべて又はいずれかが最小限になっていること」とした。 - 乳児血管腫の改善判定:5週後、8週後、12週後、16週後、20週後、24週後及び中止(来院)日
前回来院時の写真と比較し、毛細血管の拡張、紅斑、皮膚肥厚、軟組織の腫脹、解剖学的な境界の歪みから、乳児血管腫の状態を「改善」「不変」「悪化」の3段階で評価した。 - 乳児血管腫の面積、長径、色の算出:12週後、24週後、中止(来院)日
外部機関が乳児血管腫の面積、長径、色を算出した。
有効性
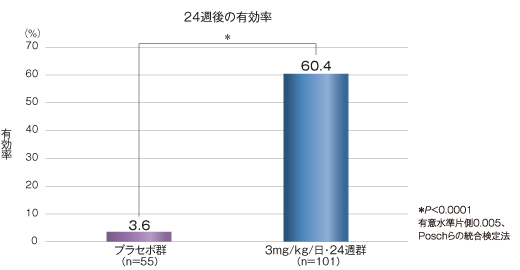
24週後における乳児血管腫に対する有効率(血管腫が「治癒又はほぼ治癒」した割合)(主要評価項目)
主解析(ステージ1+ステージ2)
24週後の乳児血管腫に対する有効率は、3mg/kg/日・24週群では60.4%であり、プラセボ群3.6%と比べて有意差が認められた(P <0.0001、Poschらの統合検定法)。
<効能・効果に関連する使用上の注意>(抜粋)
- 本剤についての十分な知識と乳児血管腫の治療経験を持つ医師が、本剤の有益性が危険性を上回ると判断した場合のみ投与すること。
本剤の承認された【用法・用量】
通常、プロプラノロールとして1日1mg/kg~3mg/kgを2回に分け、空腹時を避けて経口投与する。投与は1日1mg/kgから開始し、2日以上の間隔をあけて1mg/kgずつ増量し、1日3mg/kgで維持するが、患者の状態に応じて適宜減量する。
重要な基本的注意(抜粋)
- 初回投与時及び増量時は、小児科医との連携のもと、心拍数、血圧、呼吸状態、血糖値等を少なくとも投与2時間後まで1時間毎に確認すること。
安全性
有害事象の発現状況
(安全性解析対象集団)
発現例数(%)
| プラセボ群 (n=55) |
1mg/kg/日・ 12週群(n=98) |
1mg/kg/日・ 24週群(n=102) |
3mg/kg/日・ 12週群(n=100) |
3mg/kg/日・ 24週群(n=101) |
|
|---|---|---|---|---|---|
| 有害事象(TEAE) | 40(72.7) | 89(90.8) | 90(88.2) | 91(91.0) | 96(95.0) |
| 投与中止に至った 有害事象 |
6(10.9) | 4(4.1) | 2(2.0) | 7(7.0) | 3(3.0) |
| 因果関係が否定できない 有害事象(TEAE) |
16(29.1) | 44(44.9) | 33(32.4) | 35(35.0) | 35(34.7) |
| 重篤な有害事象 | 3(5.5) | 5(5.1) | 3(2.9) | 9(9.0) | 6(5.9) |
有害事象(TEAE*)を発現した被験者の割合は、プラセボ群で72.7%(40/55例)、1mg/kg/日・12週群で90.8%(89/98例)、1mg/kg/日・24週群で88.2%(90/102例)、3mg/kg/日・12週群で91.0%(91/100例)、3mg/kg/日・24週群で95.0%(96/101例)であった。
因果関係が否定できない主な有害事象(いずれかの群で発現率が5%以上)は、プラセボ群で中期不眠症、不眠症がそれぞれ5.5%(3/55例)、1mg/kg/日・12週群で睡眠障害10.2%(10/98例)、末梢冷感8.2%(8/98例)、下痢、中期不眠症、傾眠がそれぞれ5.1%(5/98例)、1mg/kg/日・24週群で末梢冷感7.8%(8/102例)、3mg/kg/日・12週群で中期不眠症7.0%(7/100例)、3mg/kg/日・24週群で末梢冷感8.9%(9/101例)、下痢7.9%(8/101例)、睡眠障害6.9%(7/101例)であった 。
死亡は認められず、重篤な有害事象は26例33件に発現した。重篤な副作用は5件で、内訳はプラセボ群で1件(状態悪化**)、1mg/kg/日・12週群で1件(第Ⅱ度房室ブロック)、3mg/kg/日・12週群で3件(状態悪化***、気管支炎、徐脈)であった。5件の重篤な副作用のうち4件は投与中止に至り、1件(気管支炎)では投与中断を要したが、いずれも対症療法により、又は処置なく回復した。
- TEAE: treatment-emergent adverse event 治療下で発現した有害事象、本試験では治験薬投与中及び治験薬最終投与の5日後までに発現した有害事象とした。
- 状態悪化:乳児血管腫が増大したことで開眼できなくなった事例
- 状態悪化:潰瘍形成した事例
[乳児血管腫患者を対象とした第Ⅱ/Ⅲ相臨床試験(承認時評価資料)]
- ※
- 詳細については、製品情報一覧ページ<添付文書><インタビューフォーム>及び総合製品情報概要をご覧ください。
- ※
- 総合製品情報概要をご所望の方は、弊社MR及び製品情報センター(0120-122834)にお問い合わせください。



